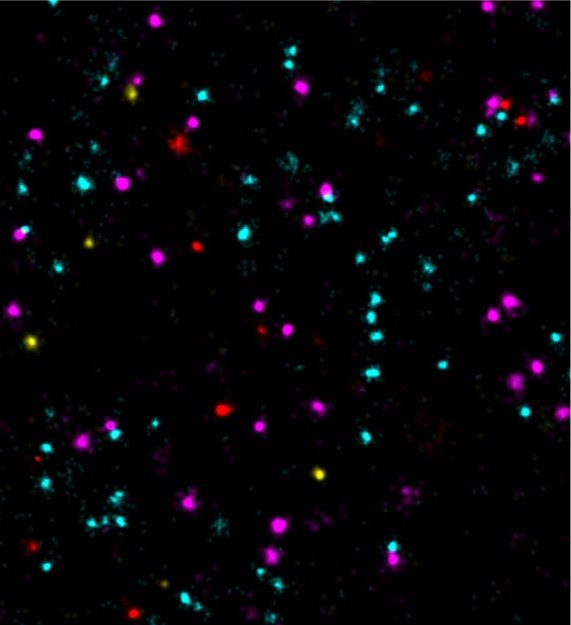
Scientists at the Max Planck Institute (MPI) of Biochemistry enable 100-times faster multiplexed DNA-PAINT microscopy using optimized DNA sequences.
• DNA-PAINT uses DNA-barcoded probes to visualize nanoscale biological structures
• Optimized DNA designs enable 100-times faster and multicolor imaging
• High throughput and molecular resolution microscopy might in the future improve our understanding of the interactions between different tumor markers
Super-resolution fluorescence microscopy can be used to visualize structures smaller than 200 nanometers, i.e. below the diffraction limit of light. One of the microscopy techniques, called DNA-PAINT, was developed by Ralf Jungmann, research group leader at the MPI of Biochemistry and Professor for Experimental Physics at the Ludwig Maximilian University Munich together with colleagues. The technique uses short ‘imagers’, dye-labeled DNA strands that temporarily bind to their target molecules in a complementary manner to produce the necessary "blinking" for super-resolution reconstruction of the images. “We have recently improved DNA-PAINT’s traditionally rather slow acquisition speed by an order of magnitude by optimizing DNA sequence design.” says Jungmann. "However, this came at the cost of losing multiplexing, which means that several structures in the cell cannot be observed simultaneously", added Jungmann. The simultaneous observation of several proteins, however, is important for the better understanding of complex signaling cascades between tumor and normal cells.”