 Congratulations on your PhD!
Congratulations on your PhD!
Geraldine Rodschinka
Identification of context-specific mRNA translation regulators in human cells by functional genomics
RG: Danny Nedialkova
Congratulations on your PhD!Identification of context-specific mRNA translation regulators in human cells by functional genomics
RG: Danny Nedialkova

Gassler, J.#, Kobayashi, W.#, Gáspár, I.#, Ruangroengkulrith, S.#, Mohanan, A., Gómez Hernández, L., Kravchenko, P., Kümmecke, M., Lalic, A., Rifel, N., Ashburn, R.J., Zaczek, M., Vallot, A., Cuenca Rico, L., Ladstätter, S., and Tachibana, K.
(#equal contribution) (IMPRS-LS students are in bold)
Science, 2022, 378, 1305-1315.
doi: 10.1126/science.abn7478
Zygotic genome activation by the totipotency pioneer factor Nr5a2
Life begins with a switch in genetic control from the maternal to the embryonic genome during zygotic genome activation (ZGA). Despite its importance, the essential regulators of ZGA remain largely unknown in mammals. On the basis of de novo motif searches, we identified the orphan nuclear receptor Nr5a2 as a key activator of major ZGA in mouse two-cell embryos. Nr5a2 is required for progression beyond the two-cell stage. It binds to its motif within SINE B1/Alu retrotransposable elements found in cis-regulatory regions of ZGA genes. Chemical inhibition suggests that 72% of ZGA genes are regulated by Nr5a2 and potentially other orphan nuclear receptors. Nr5a2 promotes chromatin accessibility during ZGA and binds nucleosomal DNA in vitro. We conclude that Nr5a2 is an essential pioneer factor that regulates ZGA.
Schmacke, N.A., O'Duill, F., Gaidt, M.M., Szymanska, I., Kamper, J.M., Schmid-Burgk, J.L., Mädler, S.C., Mackens-Kiani, T., Kozaki, T., Chauhan, D., Nagl, D., Stafford, C.A., Harz, H., Fröhlich, A.L., Pinci, F., Ginhoux, F., Beckmann, R., Mann, M., Leonhardt, H., and Hornung, V.
(IMPRS-LS students are in bold)
Immunity 55, 2271-2284.e2277.
doi: 10.1016/j.immuni.2022.10.021
IKKβ primes inflammasome formation by recruiting NLRP3 to the trans-Golgi network
The NLRP3 inflammasome plays a central role in antimicrobial defense as well as in the context of sterile inflammatory conditions. NLRP3 activity is governed by two independent signals: the first signal primes NLRP3, rendering it responsive to the second signal, which then triggers inflammasome formation. Our understanding of how NLRP3 priming contributes to inflammasome activation remains limited. Here, we show that IKKβ, a kinase activated during priming, induces recruitment of NLRP3 to phosphatidylinositol-4-phosphate (PI4P), a phospholipid enriched on the trans-Golgi network. NEK7, a mitotic spindle kinase that had previously been thought to be indispensable for NLRP3 activation, was redundant for inflammasome formation when IKKβ recruited NLRP3 to PI4P. Studying iPSC-derived human macrophages revealed that the IKKβ-mediated NEK7-independent pathway constitutes the predominant NLRP3 priming mechanism in human myeloid cells. Our results suggest that PI4P binding represents a primed state into which NLRP3 is brought by IKKβ activity.
Congratulations on your PhD!The role of central amygdala neuron subpopulations in appetitive behaviours
RG: Rüdiger Klein
Schloesser, D.#, Lindenthal, L.#, Sauer, J., Chung, K.J., Chavakis, T., Griesser, E., Baskaran, P., Maier-Habelsberger, U., Fundel-Clemens, K., Schlotthauer, I., Watson, C.K., Swee, L.K., Igney, F., Park, J.E., Huber-Lang, M.S., Thomas, M.J., El Kasmi, K.C., and Murray, P.J.
(#equal contribution)
J Cell Biol, 2023, 222.
doi: 10.1083/jcb.202207097
Senescent cells suppress macrophage-mediated corpse removal via upregulation of the CD47-QPCT/L axis
Progressive accrual of senescent cells in aging and chronic diseases is associated with detrimental effects in tissue homeostasis. We found that senescent fibroblasts and epithelia were not only refractory to macrophage-mediated engulfment and removal, but they also paralyzed the ability of macrophages to remove bystander apoptotic corpses. Senescent cell-mediated efferocytosis suppression (SCES) was independent of the senescence-associated secretory phenotype (SASP) but instead required direct contact between macrophages and senescent cells. SCES involved augmented senescent cell expression of CD47 coinciding with increased CD47-modifying enzymes QPCT/L. SCES was reversible by interfering with the SIRPα-CD47-SHP-1 axis or QPCT/L activity. While CD47 expression increased in human and mouse senescent cells in vitro and in vivo, another ITIM-containing protein, CD24, contributed to SCES specifically in human epithelial senescent cells where it compensated for genetic deficiency in CD47. Thus, CD47 and CD24 link the pathogenic effects of senescent cells to homeostatic macrophage functions, such as efferocytosis, which we hypothesize must occur efficiently to maintain tissue homeostasis.
Pinci, F., Gaidt, M.M., Jung, C., Nagl, D., Kuut, G., and Hornung, V.
Front Immunol, 2023, 13, 1074440.
doi: 10.3389/fimmu.2022.1074440
Tumor necrosis factor is a necroptosis-associated alarmin
Necroptosis is a form of regulated cell death that can occur downstream of several immune pathways. While previous studies have shown that dysregulated necroptosis can lead to strong inflammatory responses, little is known about the identity of the endogenous molecules that trigger these responses. Using a reductionist in vitro model, we found that soluble TNF is strongly released in the context of necroptosis. On the one hand, necroptosis promotes TNF translation by inhibiting negative regulatory mechanisms acting at the post-transcriptional level. On the other hand, necroptosis markedly enhances TNF release by activating ADAM proteases. In studying TNF release at single-cell resolution, we found that TNF release triggered by necroptosis is activated in a switch-like manner that exceeds steady-state TNF processing in magnitude and speed. Although this shedding response precedes massive membrane damage, it is closely associated with lytic cell death. Further, we found that lytic cell death induction using a pore-forming toxin also triggers TNF shedding, indicating that the activation of ADAM proteases is not strictly related to the necroptotic pathway but likely associated with biophysical changes of the cell membrane upon lytic cell death. These results demonstrate that lytic cell death, particularly necroptosis, is a critical trigger for TNF release and thus qualify TNF as a necroptosis-associated alarmin.
Congratulations on your PhD!Sensitive and quantitative workflows for the system-wide and targeted study of post-translational modifications
RG: Matthias Mann
Tran, M.L., Kim, Y., and von Blume, J.
Methods Mol Biol, 2023, 2557, 583-594.
doi: 10.1007/978-1-0716-2639-9_35
Quantification of Protein Exit at the Trans-Golgi Network
With one-third of all newly synthesized proteins entering the secretory pathway, correct protein sorting is essential for cellular homeostasis. In the last three decades, researchers have developed numerous biochemical, genetic, and cell biological approaches to study protein export and sorting from the trans-Golgi network (TGN). However, accurately quantifying protein transport from one compartment to the next in the secretory pathway has been challenging. The Retention Using Selective Hooks (RUSH) system is a method that allows monitoring trafficking of a protein of interest in real time, similar to a pulse-chase experiment but without the need of radiolabeling. Accurate calculations, however, are necessary and currently lacking. Here, we combine the RUSH system with live cell imaging to quantify and calculate half lives. We exemplify our approach using a soluble secreted protein (LyzC). This system will benefit membrane trafficking researchers by adding numbers to protein export and comparing the export kinetics of different cargoes and variating conditions.
Tran, M.L., Tüshaus, J., Kim, Y., Ramazanov, B.R., Devireddy, S., Lichtenthaler, S.F., Ferguson, S.M., and von Blume, J.
(IMPRS-LS students are in bold)
Traffic, 2023, 24, 4-19.
doi: 10.1111/tra.12873
The trans-Golgi Network (TGN) sorts molecular "addresses" and sends newly synthesized proteins to their destination via vesicular transport carriers. Despite the functional significance of packaging processes at the TGN, the sorting of soluble proteins remains poorly understood. Recent research has shown that the Golgi resident protein Cab45 is a significant regulator of secretory cargo sorting at the TGN. Cab45 oligomerizes upon transient Ca2+ influx, recruits soluble cargo molecules (clients), and packs them in sphingomyelin-rich transport carriers. However, the identity of client molecules packed into Cab45 vesicles is scarce. Therefore, we used a precise and highly efficient secretome analysis technology called hiSPECs. Intriguingly, we observed that Cab45 deficient cells manifest hypersecretion of lysosomal hydrolases. Specifically, Cab45 deficient cells secrete the unprocessed precursors of prosaposin (PSAP) and progranulin (PGRN). In addition, lysosomes in these cells show an aberrant perinuclear accumulation suggesting a new role of Cab45 in lysosomal positioning. This work uncovers a yet unknown function of Cab45 in regulating lysosomal function.
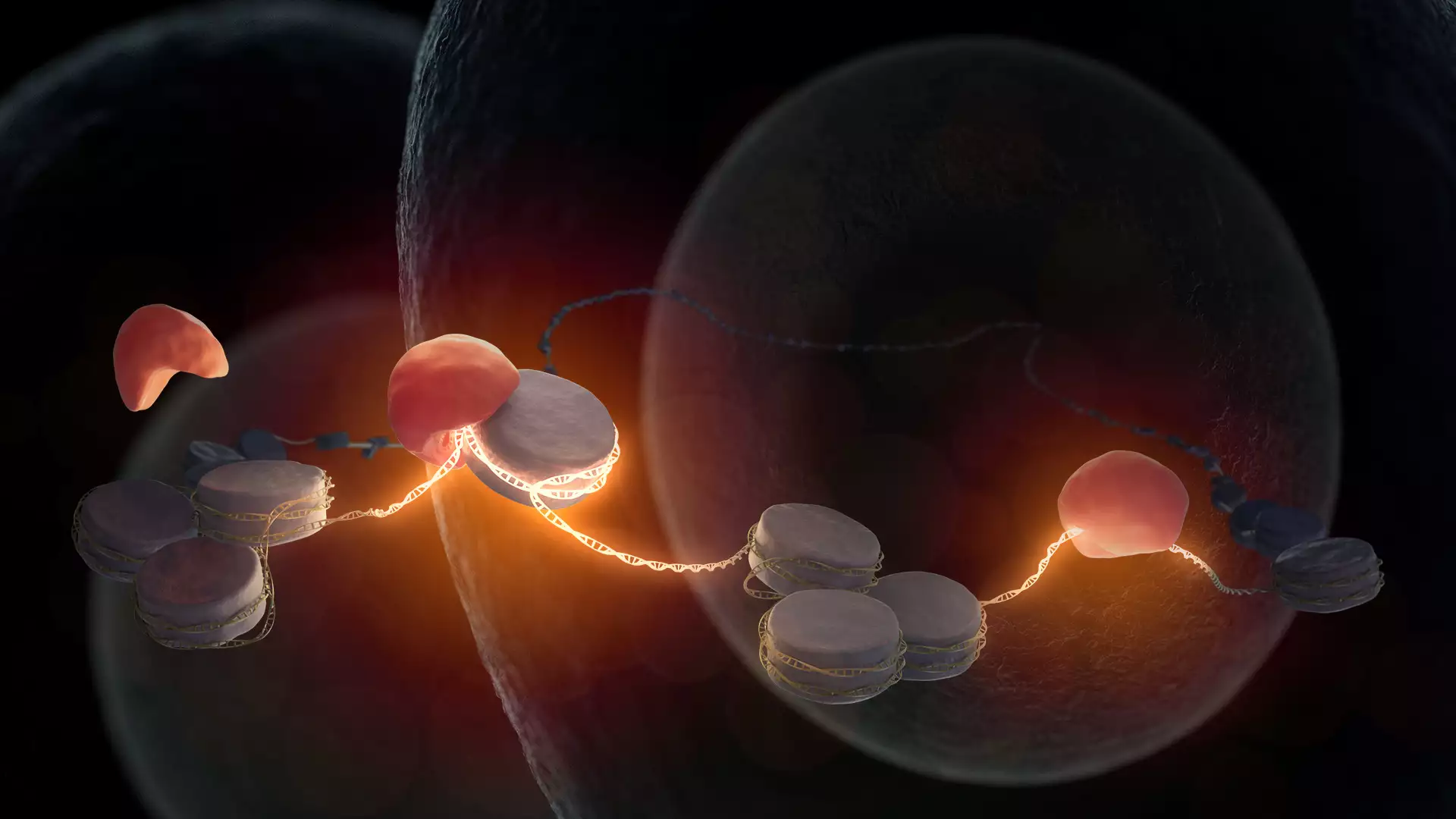
Fertilization of an egg by sperm is the beginning of new life. The maternal and paternal genetic information, which collectively store the body plan of the living being, are combined soon after fertilization. However, the DNA is still in an inactive state in the cell nucleus at this early stage of life. While the first division of the fertilized egg cell functions with the help of maternal factors stored in the egg, further development of an embryo requires access to the embryonic DNA to facilitate the synthesis of new embryonic products. It was hitherto not known which factor or factors are required for opening of DNA in mammalian embryos. Kikuë Tachibana and her team at the Max Planck Institute of Biochemistry (MPIB) have now discovered that pioneer factor Nr5a2 awakens the embryonic DNA. The research results were published in the journal Science.
The beginning of life is a fascinating process in biology. The female egg cell is fertilized through fusion with the male sperm cell. The resulting single-cell embryo forms the basis for the development of an entire organism. What molecular processes take place on the DNA of a fertilized egg to enable this special cell to have the potential to generate a new organism? Kikuë Tachibana, director at MPIB and head of the research department ‘Totipotency’, is investigating this question together with her research team using the mouse model.