 Congratulations on your PhD!
Congratulations on your PhD!
Daan Verhagen
A novel perspective on the in vivo chromatin landscape of Saccharomyces Cerevisiae
RG: Felix Müller-Planitz
Congratulations on your PhD!
A novel perspective on the in vivo chromatin landscape of Saccharomyces Cerevisiae
RG: Felix Müller-Planitz
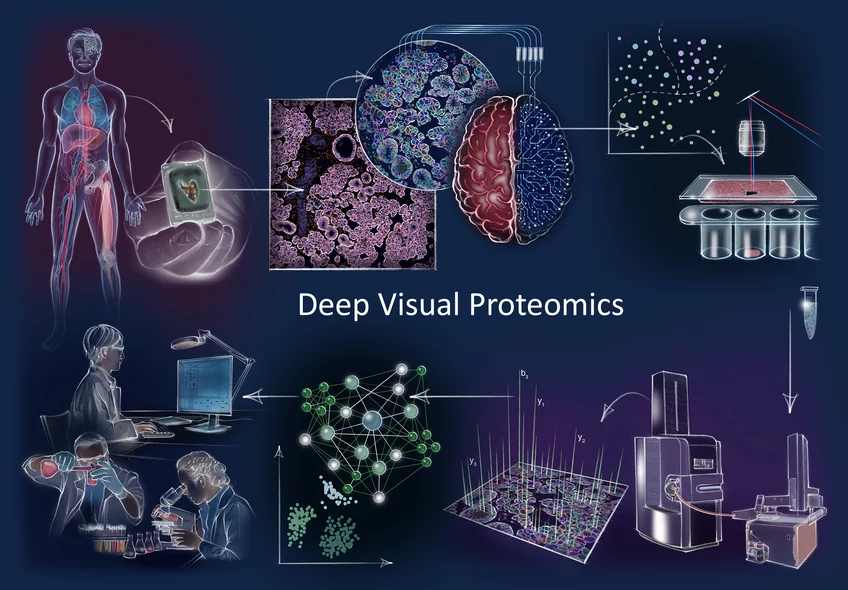
How does cancer arise? How does cellular composition influence tumor malignancy? These questions are profound and challenging to answer, but are crucial to understand the disease and find the right cure. Now, a German-Danish team led by Professor Matthias Mann has developed a ground-breaking technology called ‘Deep Visual Proteomics’. This method provides researchers and clinicians with a protein read-out to understand cancer at single cell-type resolution. The technology was published in the journal Nature Biotechnology and demonstrates its potential in a first application to cancer cells.
Proteins are among the most important players in a variety of diseases. Aptly referred to as the 'molecular workhorses of the cell’, their proper function often determines the fitness of a cell and that of an individual by extension. Matthias Mann explains: “When something goes wrong inside our cells and we become sick, you can be sure that proteins are involved in a wide range of different ways. Because of this, mapping the protein landscape can help us determine why a tumor could develop in a particular patient, what vulnerabilities that tumor has and also what treatment strategy might prove the most beneficial.”
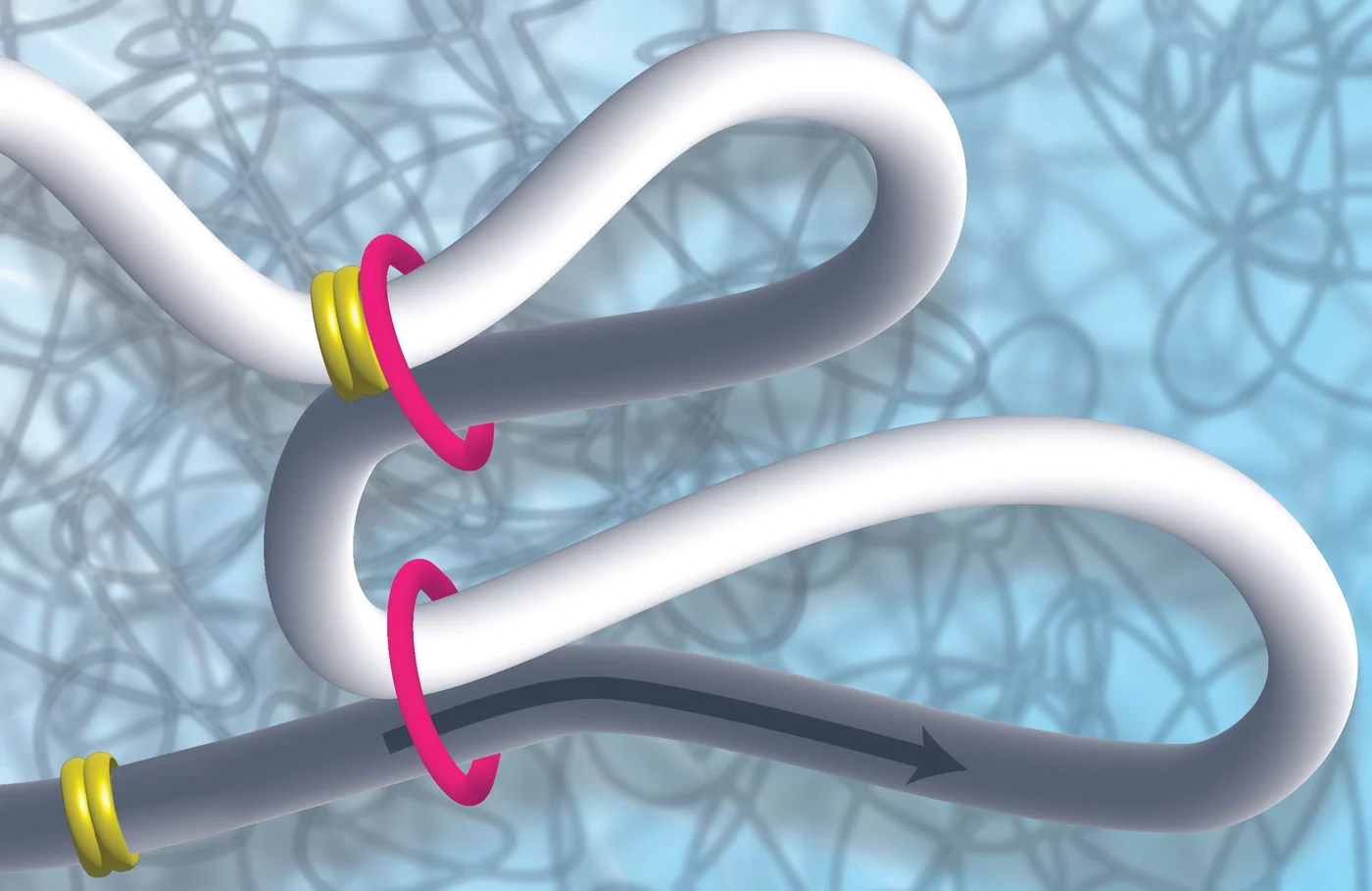
The entire genomic material of a cell must be packed into a tiny cell nucleus in such a way, that on the one hand, it can be stored in an organized manner and, on the other hand, it can be transcribed, duplicated or repaired as needed. Different proteins are responsible for space-saving packaging, which can roll up or loop the DNA. Scientists Kikuë Tachibana and Karl Duderstadt from the Max Planck Institute of Biochemistry (MPIB) in Martinsried are investigating the exact task and function of these molecular machines. They discovered that the MCM complex plays an important role in restricting DNA loop formation and thus in the three-dimensional structure of the genome and in gene regulation. The research results were published in the scientific journal Nature.
A DNA molecule is about two meters long and still has to be packed into a tiny cell nucleus. A cell nucleus is about the size of a toner particle from a printer or a fine dust particle. How does it work? How can the genetic information be stored and packaged on the one hand, but read on the other? How is it put into loops? Packaging and unpacking are also dynamic processes that must run quickly and smoothly.
Now Kikuë Tachibana, new director of the department “Totipotency” at MPIB, and her team discovered that a protein complex well known for its function in DNA replication has an unexpected role in genome folding. “During a symposium at MPIB, it emerged that my new colleague Karl Duderstadt and I shared a common interest. We decided to join forces to use complementary approaches to investigate these initial observations at a mechanistic level”. Karl Duderstadt is head of the research group "Structure and Dynamics of Molecular Machines".

Jocher, G., Grass, V., Tschirner, S.K., Riepler, L., Breimann, S., Kaya, T., Oelsner, M., Hamad, M.S., Hofmann, L.I., Blobel, C.P., Schmidt-Weber, C.B., Gokce, O., Jakwerth, C.A., Trimpert, J., Kimpel, J., Pichlmair, A., and Lichtenthaler, S.F.
EMBO reports, 2022, e54305.
doi: 10.15252/embr.202154305
ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion
The severe-acute-respiratory-syndrome-coronavirus-2 (SARS-CoV-2) is the causative agent of COVID-19, but host cell factors contributing to COVID-19 pathogenesis remain only partly understood. We identify the host metalloprotease ADAM17 as a facilitator of SARS-CoV-2 cell entry and the metalloprotease ADAM10 as a host factor required for lung cell syncytia formation, a hallmark of COVID-19 pathology. ADAM10 and ADAM17, which are broadly expressed in the human lung, cleave the SARS-CoV-2 spike protein (S) in vitro, indicating that ADAM10 and ADAM17 contribute to the priming of S, an essential step for viral entry and cell fusion. ADAM protease-targeted inhibitors severely impair lung cell infection by the SARS-CoV-2 variants of concern alpha, beta, delta, and omicron and also reduce SARS-CoV-2 infection of primary human lung cells in a TMPRSS2 protease-independent manner. Our study establishes ADAM10 and ADAM17 as host cell factors for viral entry and syncytia formation and defines both proteases as potential targets for antiviral drug development.
Congratulations on your PhD!
Mechanistic studies of GID/CTLH E3 ubiquitin ligases
RG: Brenda Schulman
Hees, J.T., and Harbauer, A.B.
Methods Mol Biol, 2022, 2431, 225-237.
doi: 10.1007/978-1-0716-1990-2_11
Live-Cell Imaging of RNA Transport in Axons of Cultured Primary Neurons
The use of fluorescent proteins has revolutionized the study of protein localization and transport. However, the visualization of other molecules and specifically RNA during live-cell imaging remains challenging. In this chapter, we provide guidance to the available methods, their advantages and drawbacks as well as provide a detailed protocol for the detection of RNA transport using the MS2/PP7-split-Venus system for background-free RNA imaging.

Elena Conti, director of the department "Cellular Structural Biology" and F. Ulrich Hartl, director of the research department "Cellular Biochemistry" at the Max Planck Institute of Biochemistry in Martinsried, near Munich, Germany, each receive an ERC Advanced grant from the European Research Council (ERC) worth 2.1 million euros. Elena Conti and her research group will investigate so-called messenger ribonucleoprotein particles in the GOVERNA project. These large complexes contain mRNA and diverse proteins. F. Ulrich Hartl and his team in the INSITUFOLD project will investigate the basic dynamic functions of chaperones and their interactions with clients in living cells in real time.
GOVERNA Project
Messenger ribonucleic acids, known as mRNAs, contain the genetic information that determines the structure of proteins to be produced. They are read from the DNA in the cell nucleus and passed on to the protein synthesis machinery in the cell plasma, the place where the mRNA is read and translated into amino acid chains. During their production, different proteins bind to the mRNA strands to form complexes called messenger ribonucleoprotein particles, mRNPs. Over the past few decades, Elena Conti and her team have deciphered the complex assembly of large, central machinery such as the exosome, a protein structure that degrades mRNAs. In her current project, GOVERNA, she will use state-of-the-art technology to explore the three-dimensional assembly of a wide range of other proteins that interact with mRNAs in dynamic and diverse ways. Knowledge of their structure their interplay and functioning will help understand the basic life cycles of mRNAs in cells. Given the recent developments in mRNA-based therapeutics, the study of mRNPs is particularly timely and relevant.

"If you're going on a long journey, it's better to pack light and pack smart," says Angelika Harbauer, head of the research group Neurometabolism at the Max Planck Institute for Biological Intelligence, in foundation (i.f.). The sentence summarizes the motto by which cellular power plants known as mitochondria travel through the long extensions of nerve cells. Harbauer's research shows that, instead of a protein that is important to them, travelling mitochondria take along the blueprints needed to produce that protein. The results were published in the journal Neuron and have now been highlighted in a feature article in the journal Autophagy.
Proteins are biological machines that perform a wide variety of tasks. In one of her research projects, which started at Boston Children's Hospital in Tom Schwarz's group, Angelika Harbauer studies the production of one of these proteins in more detail. The PINK1 protein is found in mitochondria, the power plants of the cell, and ensures that defective mitochondria are sorted out and recycled. This prevents the cell from being damaged by defective mitochondria. PINK1 is needed wherever there are mitochondria.
Congratulations on your PhD!
Cryo-EM structure of CUL5-ARIH2 E3-E3 ligase complex reveals novel allosteric NEDD8 mechanism
RG: Brenda Schulman
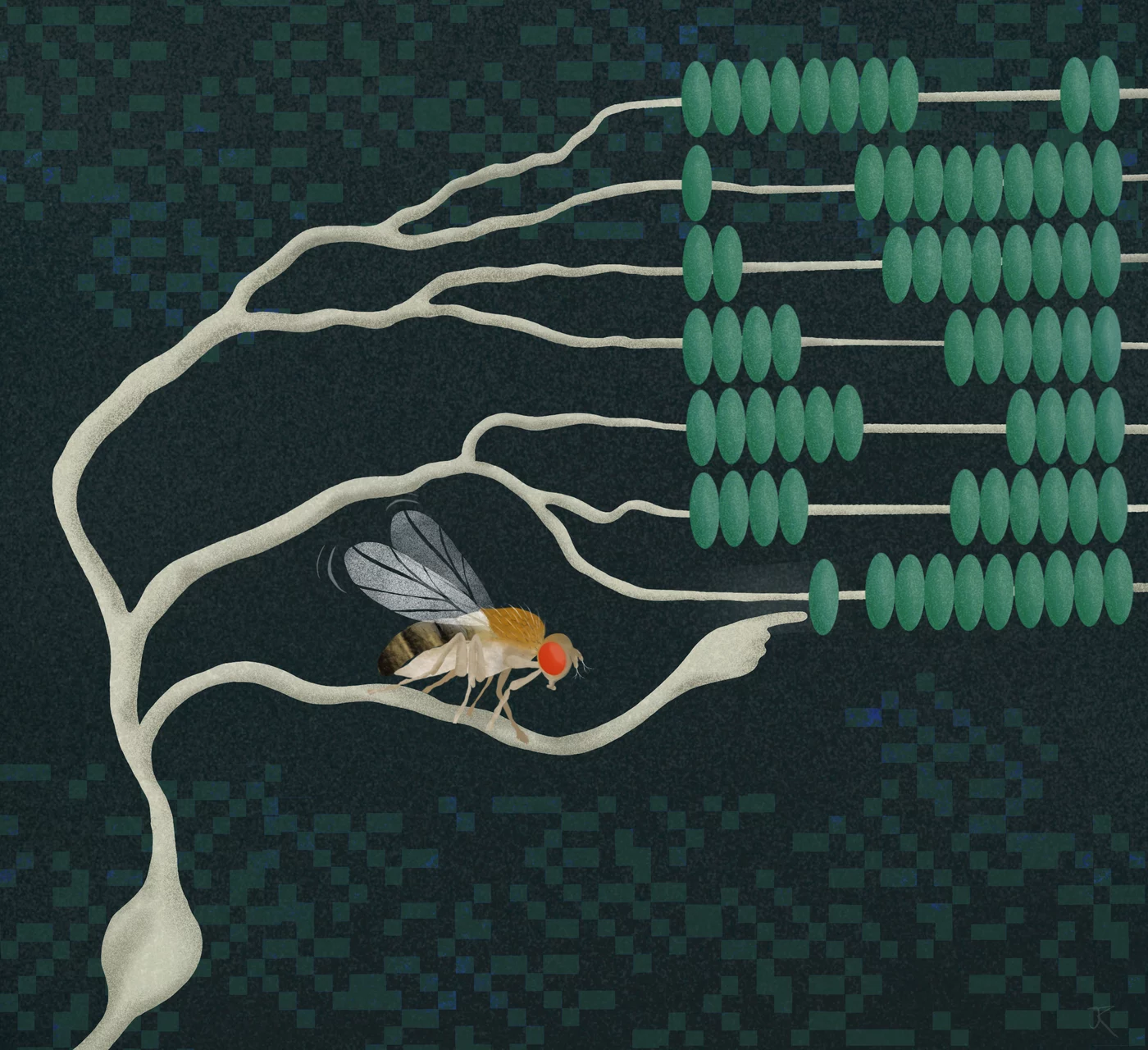
Neurons are constantly performing complex calculations to process sensory information and infer the state of the environment. For example, to localize a sound or to recognize the direction of visual motion, individual neurons are thought to multiply two signals. However, how such a computation is carried out has been a mystery for decades. Researchers at the Max Planck Institute for Biological Intelligence, in foundation (i.f.), have now discovered in fruit flies the biophysical basis that enables a specific type of neuron to multiply two incoming signals. This provides fundamental insights into the algebra of neurons – the computations that may underlie countless processes in the brain.
We easily recognize objects and the direction in which they move. The brain calculates this information based on local changes in light intensity detected by our retina. The calculations occur at the level of individual neurons. But what does it mean when neurons calculate? In a network of communicating nerve cells, each cell must calculate its outgoing signal based on a multitude of incoming signals. Certain types of signal will increase and others will reduce the outgoing signal – processes that neuroscientists refer to as 'excitation' and 'inhibition'.